COSMO¶
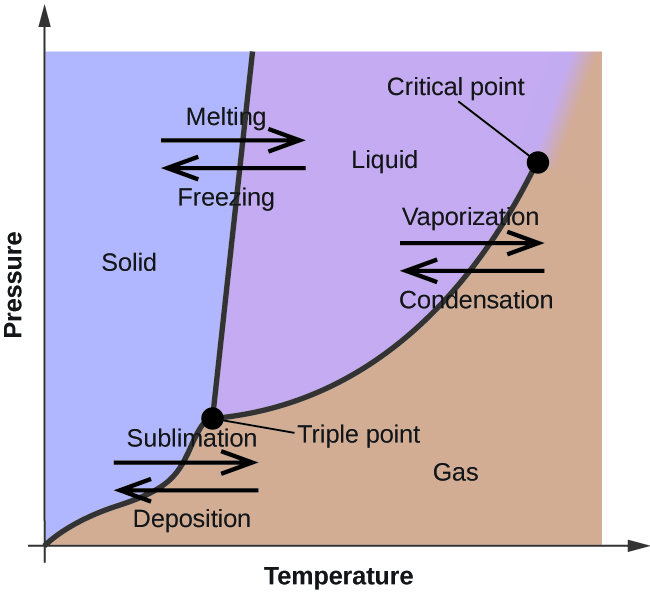
Figure 1: Temperature-Pressure phase diagram 1
Introduction to thermodynamics¶
The chemical potential is an important theoretical quantity in the field of thermodynamics and describes the change in energy of a chemical system of multiple components where the composition of a system is under change.
The composition of atoms or molecules in the system can change due to a reaction or due to a flux between the system/phase boundaries. Common examples of such chemical phenomena can be observed in our environment or in industrial processes (Figure 1).
The chemical potential can be related to thermodynamic state functions, namely internal energy \(U(S,V)\), enthalpy \(H(S,p)\), Helmholtz energy \(F(T,V)\), Gibbs enery \(G(T,p)\):
and one can derive that:
Phase equilibria calculations such as vapour liquid equilibria (VLE), liquid liquid equilibria (LLE), solid liquid equilibria (SLE) are states where the chemical potentials of all the phases are equal:
With this quick recap of some basic thermodynamic equations we will move on to discuss the theory of implicit solvation models and circle back to the connection between solvation models and thermodynamic properties in the end of the solvation models section.
Solvation models¶
When considering the environment of an atom or molecule in solution, then the electronic effect of the atoms/molecules surrounding an atom/molecule of interest needs to be taken into account.
Implicit solvation models describe this interaction between a solute and a solvent with considering a polarizable continuum (also called dielectric medium). This electronic spatial profile of molecules allows to map a profile of positive and negative charge distributions on a specific surface around the solute molecule. This profile is called the apparent surface charge (ASC) density \(\sigma(\vec{s})\) and defines a border between the solute and the continuum of the solvent. The polarity of the solvent will have an influence on the ASC density and the polarity is defined with the dielectric constant \(\epsilon(\vec{r})\).
The electrostatic solvation energy of a molecule in solutions is defined as:
and is the change in energy when transferring the molecule under consideration from the gas phase into the solution/solvent(s) phase. The integration is performed over the solute surface points \(s\).
To retrieve the before mentioned apparent surface charge (ASC) density \(\sigma(\vec{r})\) of the solute molecule, the Poisson equation has to be solved 2:
Where the equation simplifies with \(\epsilon(r) = 1\) within the solute molecule cavity to:
and with \(\epsilon(r) = \epsilon\) (set to a constant value, e.g. to 80 for water) outside of the solute cavity to:
There exist different ways how the Poisson equation can be solved where the electrostatic surface around the molecule is dissected into smaller pieces (tesserae). The Poisson equation is then being solved in combination with a tesserae dissection routine.
In this article we present the DPCM, CPCM/COSMO and IPCM/IEFPCM models which differ based on the implemented boundary conditions. Important to highlight is that the DPCM model relies on the net electric field, \(E_n\), and the COSMO and IEFPCM models depend only on the net potential (\(\phi(s)\)) at the cavity surface of a given solute molecule . IEFPCM reduces to the COSMO model when \(\epsilon\) approaches infinity [4].
To solve the Poisson equation numerically and to provide a general mathematical formulation, one can recast the equation in matrix form:
with \(\vec{q}\) storing the unknown cavity surface charges, \(\mathbf{K}\) (the solvent response matrix) being a square matrix storing the information about the geometry (tesserae location, area) and dielectric information of the medium.
\(\mathbf{K}\) is defined by the matrices/operators \(\mathbf{A}\), \(\mathbf{D}\) and \(\mathbf{S}\) presented later. \(\vec{f}\) holds the normal component of the electrostatic quantity at the tesserae. Tomasi et al. 3 provide a nice overview (Table 1 in the publication) how this equation in matrix form can be utilized to calculate the ASC density with various methods (e.g. PCM/DPCM, CPCM/COSMO, IEFPCM).
The DPCM ASC density \(\sigma(\vec{s})\) defintion with the exact dielectric boundary condition (EDBC) at the interface (surface) of the cavity, with \(\vec{s}\) being a point on the cavity surface, is:
By applying the COSMO boundary conditions (COSMO-BC) to solve the Poisson equation, one obtains the CPCM/COSMO based ASC density equation:
And the IEFPCM boundary condition leads to the following equation of the ASC density:
with \(\mathbf{I}\) being the identity matrix, \(\mathbf{A}\) is the symmetric Coulomb interaction matrix (COSMO matrix) of the charges on the tesserae/segments, \(\mathbf{D}\) and \(\mathbf{S}\) are components of the Calderon projector 4 where \(\mathbf{D}\) is the non-symmetric matrix of the normal electric field components generated by the screening charges and \(\mathbf{S}\) is a diagonal matrix of the segment areas.
A scaling function \(f(\epsilon)\) is applied in the COSMO model to re-scale the unscreened charge density \(\sigma^*\) (\(\epsilon \to \infty\)) via:
\(x\) is an empirical parameter set to 0.5 for neutral solutes and 0 for ions.
Klamt et al. 5 also showed that the prediction performance of IEFPCM and COSMO is near to similar if the correct scaling factor is chosen. The beauty of the COSMO model is its computational effectiveness and robustness as it does not need to take into account the D-matrix 5. The so called sigma-profile \(p(\sigma)\) (a histogram) which is generated from the ASC density \(\sigma(s)\) can then be considered as a look up data table for each molecule to calculate thermodynamic properties for temperature-dependent multi-component systems such as
- Thermodynamic equilibria diagrams: vapour-liquid (VLE), solid-liquid (SLE), liquid-liquid (LLE)
- Solubility of a solute in solvent systems
- Partition coefficient of a compound between two liquid phases
The connecting quantity between these properties and \(p(\sigma)\) is the chemical potential and we will present in one of the next blog articles further explanation how the set of equations can be derived to calculate solubility etc.
Application programming interface (API)¶
The full documentation of the API endpoints is available in the Cebule Engine Dashboard when you have subscribed to the Cebule Tier.
 Figure 4: Documentation page of API with all the endpoints and direct data entry possibilities to test the endpoints and to initiate calculation tasks.
Figure 4: Documentation page of API with all the endpoints and direct data entry possibilities to test the endpoints and to initiate calculation tasks.
The general API structure can be categorized with the following three endpoints:
POST request via endpoint /api/tasks¶
The POST request holds the following needed data:
- name of the calculation task
- basis set
- dielectric constant
- molecule geometry in xyz coordinates
- the quantum chemical approximation method, for example SCF
- the van der Waals radii of the individual atoms
- and the element definition for the individual atoms.
In this way you are able to send multiple calculation tasks to the high performance COSMO algorithm running on energy efficient Arm processors. Intel, AMD and Nvidia processors are also supported.
GET request /api/tasks¶
A GET request via the /api/tasks endpoint will always deliver a list of all tasks and the status of the individual task.
The list of dictionaries with each individual task has the following schema:
One can also retrieve the list of tasks via Python with the following code snippet:
GET request /api/tasks/{task_id}¶
By sending a GET request to this endpoint with the specific task_id, one receives the information of either a successful response (Code 200) or an error response (Code 422).
The schema looks as follows for the successful response:
And the schema for the error response is:
The following code snippet shows how to send a request via Python:
S3 buckets¶
All tasks and results data are stored in a S3 bucket and can be directly accessed through the cloud provider S3 bucket page (see Figure 5) or one can directly request the individual tasks' data via this Python snippet for an AWS S3 bucket (the boto3 package has to be installed):
YOUR_OBJECT_KEY has the following path: "tasks/TASK_ID/TASK_NAME_cosmo.json"
One can also make use of the AWS CLI via the command line:
If no TASK_NAME was defined in the input then the results can be retrieved as follows:
In the AWS dashboard the individual tasks folder view can be seen in Figure 5.
 Figure 5: S3 bucket view of all task folders for specific S3 bucket. The bucket name is defined by the user on the cloud provider platform of choice.
Figure 5: S3 bucket view of all task folders for specific S3 bucket. The bucket name is defined by the user on the cloud provider platform of choice.
COSMO results data¶
You can get full access to all variables and parameters in the COSMO model. The results include for example:
- the number of cosmo segments and their coordinates
- the molecular surface area
- the molecular COSMO volume
- the cosmo surface charges for each segment
When retrieving the results of your calculation tasks you can get a complete and extensive overview which data is currently retrievable from the high performance COSMO implementation.
How to send off a task for a COSMO and sigma-profiles calculation via the Python SDK¶
The following code snippet shows how Cebule can be used to perform a COSMO calculation with the Python SDK:
from dotenv import load_dotenv
import mqsdk
from mqsdk.core.cebule import TaskType
import os
# Initialize the SDK with user credentials
load_dotenv()
session = mqsdk.Cebule(os.environ['EMAIL'], os.environ['PASSWORD'])
input_data = {
"geometry": [
0.0, 0.0000, -0.1294,
0.0, -1.4941, 1.0274,
0.0, 1.4941, 1.0274
],
"symbols": ["O", "H", "H"],
"method": "SCF",
"basis": "6-31g**",
"dielec": 78.0,
"radius": [1.40,1.16,1.16]
}
task = session.cebule.create_task("COSMO Water Test", TaskType.COSMO,
input_data, max_processors=16)
print(task)
Find the whole Jupyter notebook under the following link:
https://gitlab.com/mqsdk/python-sdk/-/blob/main/notebooks/2_COSMO_task_Cebule.ipynb
-
Openstax - chemistry 2e, 10.4 phase diagrams. \url https://openstax.org/books/chemistry-2e/pages/10-4-phase-diagrams. Accessed: 2024-10-27. ↩
-
John David Jackson. Classical electrodynamics. Wiley, 3rd ed. edition, 1999. ISBN 9780471309321. ↩
-
Jacopo Tomasi, Benedetta Mennucci, and Roberto Cammi. Quantum mechanical continuum solvation models. Chemical Reviews, 105(8):2999–3094, 2005. doi:10.1021/cr9904009. ↩
-
W. Hackbusch. Integral equations :. Volume 120. Birkhäuser,, 1995. ISBN 0817628711, 3764328711, 9780817628710, 9783764328719. ↩
-
A. Klamt, C. Moya, and J. Palomar. A comprehensive comparison of the iefpcm and ss(v)pe continuum solvation methods with the cosmo approach. Journal of Chemical Theory and Computation, 11(9):4220–4225, 2015. doi:10.1021/acs.jctc.5b00601. ↩↩